Bạch cầu kinh dòng Lympho
Bệnh bạch cầu kinh dòng lympho (CLL) được đặc trưng bởi sự tích tụ tiến triển của các tế bào lympho B ác tính trưởng thành về mặt kiểu hình. Phần nguyên phát của bệnh bao gồm máu ngoại vi, tủy xương, lách, và hạch lympho. Các triệu chứng có thể có hoặc không bao gồm hạch to, lách to, gan to, mệt mỏi, sốt, đổ mồ hôi ban đêm, sụt cân và cảm thấy no sớm. Chẩn đoán bằng phương pháp đo dòng tế bào và type miễn dịch của máu ngoại vi. Điều trị bị trì hoãn cho đến khi các triệu chứng phát triển và thường liên quan đến hóa trị và liệu pháp miễn dịch. Tuy nhiên, các phương pháp điều trị đang được tiến hành, và phác đồ bậc một có thể bao gồm các thuốc điều trị đích như thuốc ức chế Bruton tyrosine kinase (BTk) và Bcl-2, có hoặc không có hóa trị liệu.
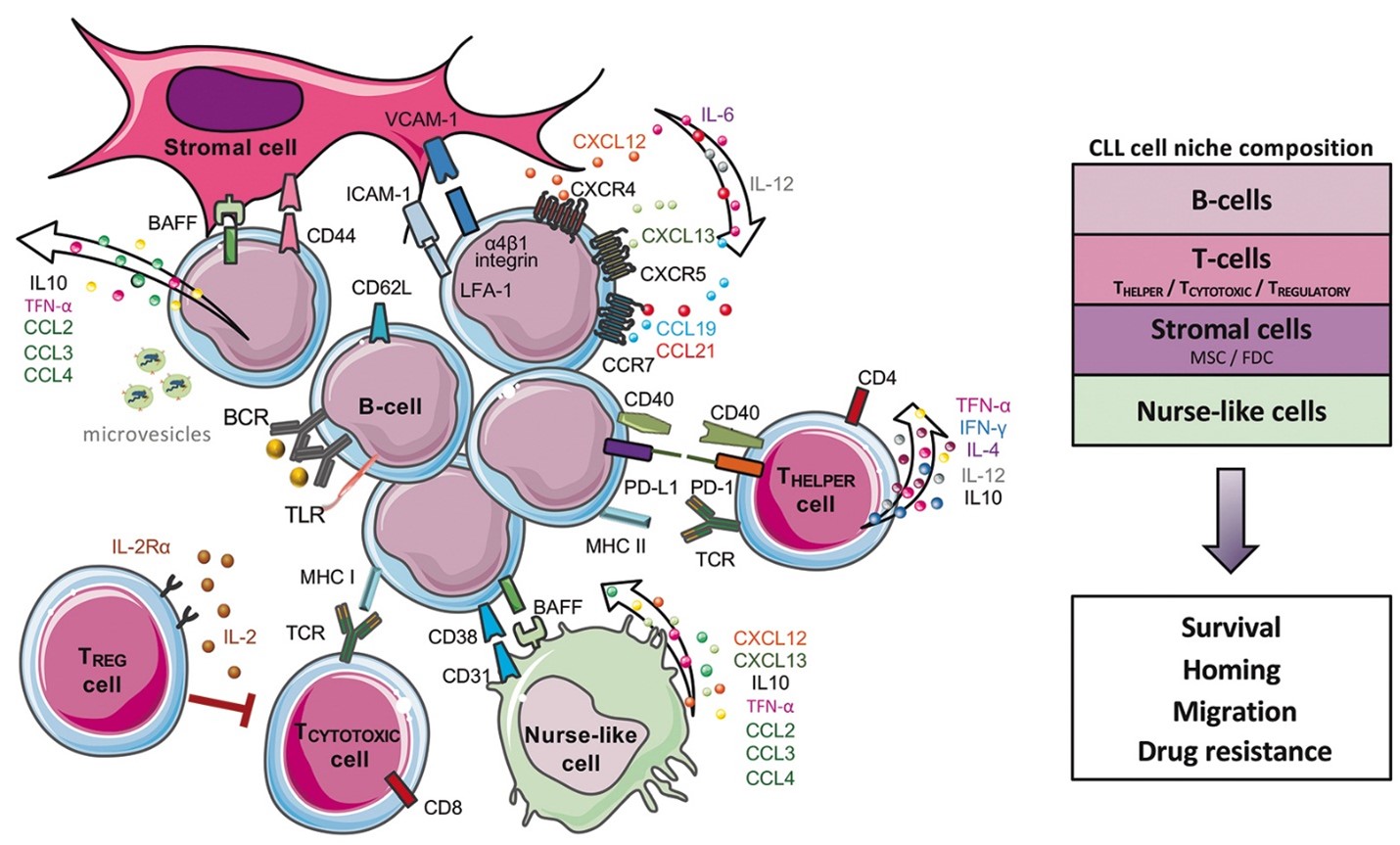
Cơ chế hình thành CLL
Trong bệnh bạch cầu mạn tính dòng lympho, tế bào CD5 + B trải qua quá trình biến đổi ác tính. Các tế bào B được kích hoạt liên tục bằng cách thu nhận các đột biến dẫn đến tăng tế bào lympho bào B (MBL). Sự tích tụ thêm các bất thường di truyền và sự chuyển đổi tế bào B đơn dòng sau đó dẫn đến CLL. Tế bào lympho ban đầu tích tụ trong tủy xương và sau đó lan đến các hạch bạch huyết và các mô lympho khác, cuối cùng gây ra lách to, gan to, và các triệu chứng toàn thân như mệt mỏi, sốt, đổ mồ hôi ban đêm, và giảm cân không chủ ý.
Ở CLL tiến triển, sự sinh máu bất thường sẽ gây thiếu máu, giảm bạch cầu trung tính, giảm tiểu cầu và giảm sản xuất globulin miễn dịch. Giảm gammaglobulin có thể gặp ở 2/3 bệnh nhân, làm tăng nguy cơ biến chứng nhiễm trùng. Bệnh nhân tăng nhạy cảm với tan máu tự miễn (với xét nghiệm kháng globulin trực tiếp dương tính) và giảm tiểu cầu tự miễn.
CLL có thể tiến triển thành bệnh bạch cầu tăng sản tế bào B và có thể chuyển sang dạng cao hơn u lympho không Hodgkin. Khoảng 2 đến 10% trường hợp CLL phát triển thành u lympho tế bào B lớn lan tỏa (gọi là chuyển đổi tế bào B).
Triệu chứng và dấu hiệu của CLL
Bệnh nhân thường không triệu chứng giai đoạn sớm, với những triệu chứng không đặc hiệu (như mệt mỏi, suy yếu, chán ăn, sụt cân, sốt, đổ mồ hôi ban đêm), có thể dẫn đến việc làm xét nghiệm. Hơn 50% bệnh nhân có hạch to. Hạch to có thể khu trú (với hạch vùng cổ và hạch thường gặp nhất) hoặc toàn thân. Bệnh lách và gan to ít gặp hơn bệnh bạch cầu. Liên quan đến da (lớp biểu bì bệnh bạch cầu) là rất hiếm.
Chẩn đoán CLL
- CBC và phết máu ngoại vi
- Đo tế bào dòng chảy của máu ngoại vi
- Kiểu hình miễn dịch
Chẩn đoán bệnh bạch cầu mạn tính dòng lympho lần đầu tiên được nghi ngờ khi có tăng tế bào lympho ngoại biên > 5000/mcL (> 5 × 109/L) được tìm thấy. Xét nghiệm tế bào máu ngoại vi có thể xác định được dòng tế bào B trong máu. Các lympho bào lưu hành nên biểu hiện các chuỗi nhẹ CD5, CD19, CD20, CD23, và kappa hoặc lambda. Bệnh nhân có số lượng tế bào lympho tuyệt đối 5000/mcL (< 5 × 109/L) nhưng bằng chứng của dòng được chẩn đoán với lymphocytosis tế bào B đơn dòng (MBL). Khoảng 1 đến 2% trường hợp tế bào lympho bào B đơn dòng tiến triển đến CLL mỗi năm. Chọc hút dịch tủy xương và sinh thiết không cần thiết để chẩn đoán CLL. Tuy nhiên, nếu được thực hiện, tủy thường cho thấy > 30% tế bào lympho.
Các dấu hiệu khác khi chẩn đoán có thể bao gồm giảm gammaglobulin (< 15% trường hợp) tăng lactate dehydrogenase (LDH), tăng axit uric, tăng men gan, và hiếm khi tăng canxi huyết. Các xét nghiệm di truyền tế bào và phân tử được thực hiện từ máu ngoại vi tại thời điểm chẩn đoán giúp xác định tiên lượng.
Phân loại sử dụng hệ thống phân loại Rai hoặc Binet. Cả hai hệ thống đều không tiên đoán được sự tiến triển của bệnh. Hình ảnh thường quy không được khuyến cáo cho giai đoạn ban đầu.Tiên lượng về CLL
Tiền sử tự nhiên của bệnh bạch cầu lymphocytic mạn tính rất khác nhau. Thời gian sống thêm khoảng từ 2 đến > 20 năm, trung bình khoảng 10 năm. Bệnh nhân ở giai đoạn từ 0 đến II của Rai sống từ 5 đến 20 năm mà không điều trị.
Các tính năng tiên lượng khác của CLL bao gồm
- Thời gian nhân đôi của tế bào lympho
- Bất thường về di truyền
Thời gian nhân đôi số tế bào lympho là số tháng cần số lượng tế bào lympho tuyệt đối tăng gấp đôi. Bệnh nhân không được điều trị với thời gian nhân đôi số tế bào lympho < 12 tháng có giai đoạn lâm sàng tích cực hơn.
Các bất thường tế bào có nguy cơ cao bao gồm del (17p) và del (11q). Các đặc điểm tiên lượng bất lợi khác bao gồm một gen biến thể globulin miễn dịch không biến đổi, sự hiện diện của CD38 trên tế bào học dòng chảy, và biểu hiện của ZAP-70.
Điều trị CLL
- Liệu pháp miễn dịch, liệu pháp đích và xạ trị
- Chăm sóc hỗ trợ
Bệnh bạch cầu mạn tính dòng lympho được coi là không thể chữa được với tiêu chuẩn chăm sóc hiện tại; điều trị nhằm mục đích cải thiện triệu chứng. Điều trị sốt phát ban do Rickettsia lựa chọn một trong các phác đồ sau:
- Các triệu chứng do CLL
- Tăng tế bào lympho tiến triển với tăng ≥ 50% trong khoảng thời gian 2 tháng
- Thời gian nhân đôi của tế bào lympho dưới 6 tháng
Các triệu chứng được điều trị kịp thời ở bệnh nhân CLL bao gồm:
- Các triệu chứng toàn thân (sốt, đổ mồ hôi ban đêm, mệt mỏi cực độ, sút cân)
- Gan to, lách to, hoặc hạch to
- Nhiễm trùng tái phát
- Di căn tủy xương có thể gây thiếu máu và/hoặc giảm tiểu cầu.
Điều trị bệnh các lựa chọn bao gồm:
- Liệu pháp hóa trị liệu
- Mục tiêu
- Liệu pháp bức xạ
Chăm sóc hỗ trợ bao gồm:
- Truyền các tế bào hồng cầu đóng gói cho bệnh thiếu máu
- Truyền tiểu cầu nếu chảy máu do giảm tiểu cầu
- Điều trị kháng sinh khi nhiễm khuẩn, nhiễm nấm hoặc virus
Vì giảm bạch cầu trung tính và hạ gammaglobulin miễn dịch nên hạn chế khả năng giết vi khuẩn, cần điều trị kháng sinh sát khuẩn. Truyền gamma-globulin nên được xem xét để điều trị cho bệnh nhân bị hạ đường huyết và nhiễm trùng khó chữa hoặc để dự phòng khi xảy ra.≥ ≥ 2 trường hợp nhiễm trùng nặng trong vòng 6 tháng.
Liệu pháp ban đầu
Hóa trị liệu mục đích là để
- Giảm các triệu chứng cấp tính
- Phục hồi lâu dài
- Kéo dài sự sống
Không có phác đồ điều trị bằng hóa chất tiêu chuẩn. Lựa chọn điều trị ban đầu phụ thuộc vào đặc điểm của bệnh nhân, đặc điểm của bệnh như sự hiện diện của del (17p) và các mục tiêu bao quát của điều trị.
CLL tái phát hoặc kháng trị
CLL tái phát hoặc kháng trị nên được xác nhận về mặt mô học trước khi bắt đầu điều trị. Chuyển đổi sang u lympho tế bào lớn (chuyển đổi của Richter) cần được loại trừ một cách cụ thể. Bệnh nhân không triệu chứng với CLL tái phát được theo dõi chặt chẽ các triệu chứng cần điều trị. Các yếu tố ảnh hưởng đến điều trị bao gồm:
- Liệu pháp ban đầu được sử dụng
- Thời gian đáp ứng ban đầu
Ibrutinib (một chất ức chế Btk) có thể cải thiện tỷ lệ đáp ứng và tỷ lệ sống còn không tăng trong CLL tái phát hoặc kháng trị. Ibrutinib được tiếp tục cho đến khi độc tính phát triển hoặc bệnh tiến triển. Các liệu pháp điều trị hiệu quả khác cho CLL tái phát bao gồm idelalisib (thuốc ức chế uống phosphoinositide 3'-kinase [PI3K] delta) và venetoclax (thuốc ức chế uống Bcl-2). Venetoclax có thể được sử dụng cho những bệnh nhân có del (17p), người đã nhận được tối thiểu một liệu pháp điều trị trước đó.
Đơn trị liệu với kháng thể đơn dòng chống CD20 (rituximab, ofatumumab, obinutuzumab) có thể tạm thời giảm nhẹ triệu chứng.
Cần xem xét ghép tế bào gốc tạo máu nên cân nhắc cho những bệnh nhân vừa sức.
Liệu pháp bức xạ
Có thể chiếu xạ giảm nhẹ cho những vùng bị nổi hạch hoặc những vùng gan và lá lách không đáp ứng với hóa trị liệu. Chiếu xạ toàn thân với liều lượng nhỏ đôi khi thành công trong việc cải thiện tạm thời các triệu chứng.
Ths.BS Vương Sơn Thành – TT Huyết học – Bệnh viện Bạch Mai